metadata <- read.table(file = "https://raw.githubusercontent.com/emrahkirdok/ybva/main/data/metadata-GSE50161.txt", header = T)29 Beynin kanseri mikrodizin analizi
Bu pratik kapsamında GSE50161 numaralı çalışmadan elde edilen beyin kanseri verisi üzerine küçük bir çalışma yapılacaktır. Bu çalışma kapsamında glioblastoma ve sağlıklı, normal beyin dokularından elde edilen gen anlatım profili incelenecektir.
29.1 Veri yükleme
Örnekler hakkında metaveri bilgisini yükleyiniz:
Yüklediğiniz metaverinin sütun isimlerini elde eden kodu yazınız:
colnames(metadata)[1] "Ornek" "Durum"Çalışmada kaç tane örnek kullanılmış? Kullanılan örnek sayısını geri döndüren kodu yazınız:
length(metadata$Ornek)[1] 47Çalışmada kullanılan koşulların tablosunu oluşturan kodu yazınız:
table(metadata$Durum)
glioblastoma normal
34 13 Şimdi de gen anlatım verisini yükleyiniz:
microarray <- read.table(file = "https://raw.githubusercontent.com/emrahkirdok/ybva/main/data/brain-expression-small-GSE50161.txt", header = T)Yüklediğiniz gen anlatım verisini matrise çeviriniz:
microarray <- as.matrix(microarray)Bu matrisin satır ve sütun sayılarını geri döndürünüz:
dim(microarray)[1] 1999 47Acaba kaç tane gen incelenmiş? Bunu geri döndüren kodu yazınız:
nrow(microarray)[1] 199929.2 Verinin Kalite Kontrolü
Elinizdeki veriyi histogram oluşturarak kontrol ediniz:
hist(microarray)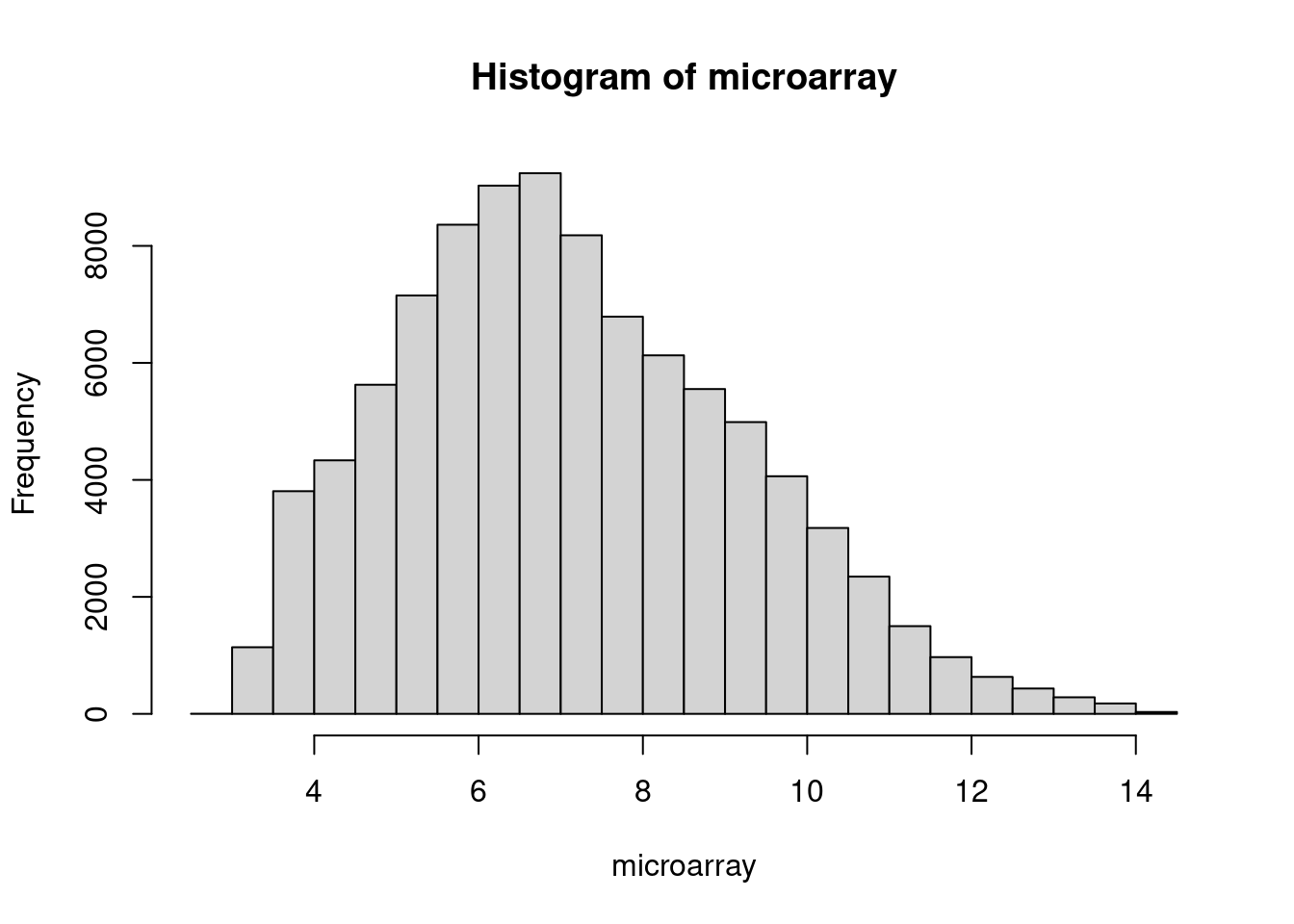
Elinizdeki verinin kutu grafiklerini oluşturunuz:
boxplot(microarray)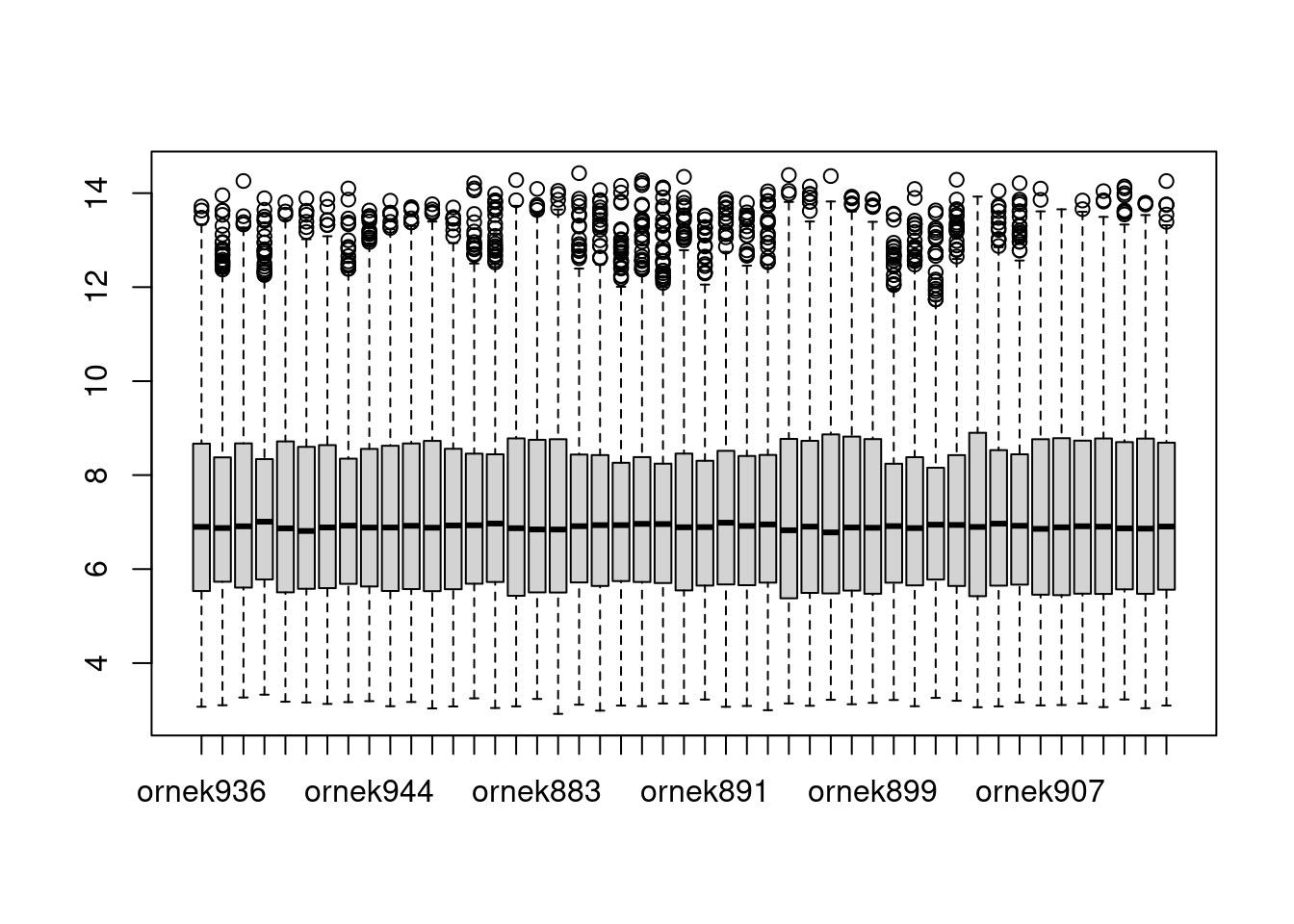
Hiyerarşik kümeleme yapınız:
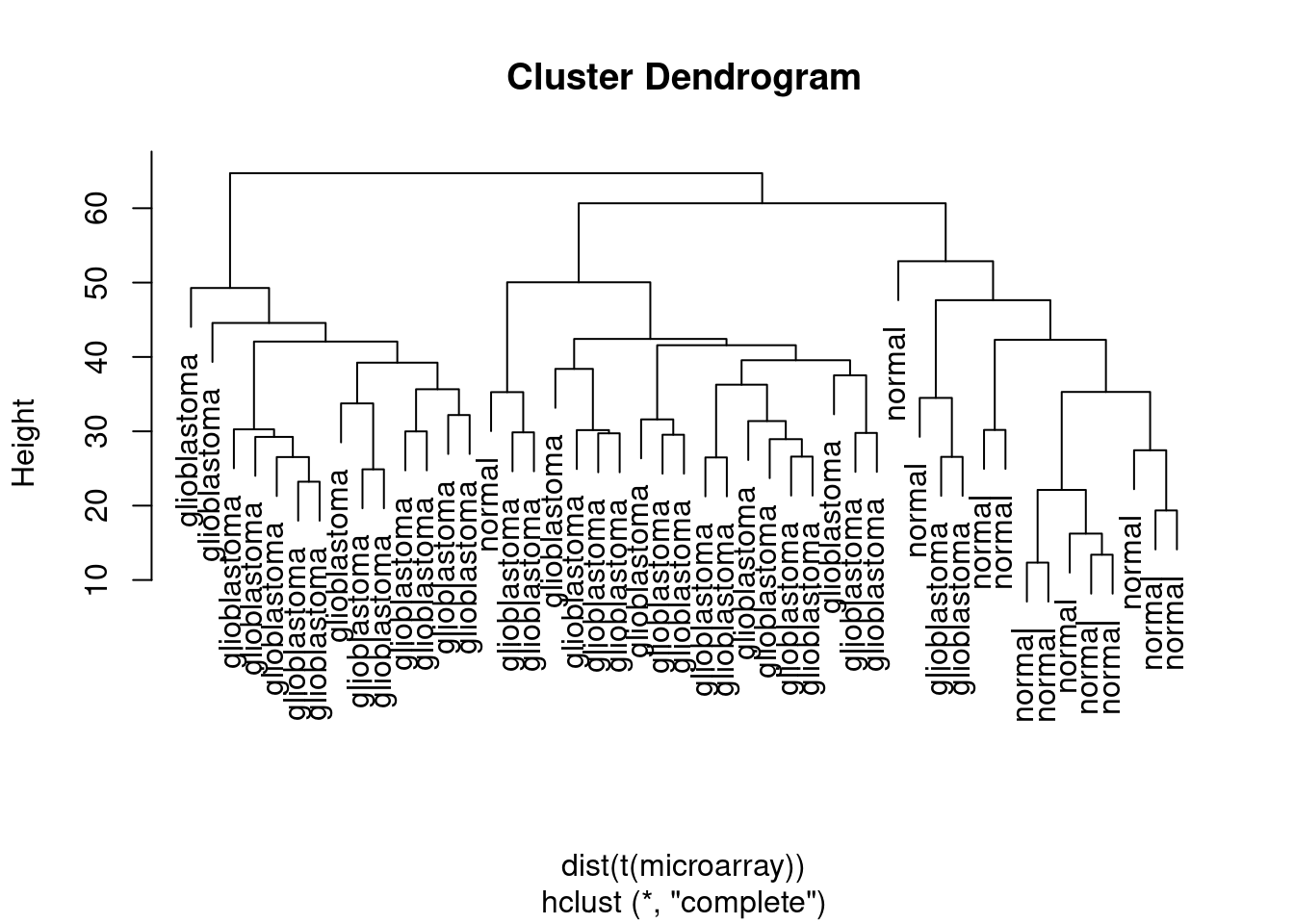
Bu bilgilere göre şu sorunun cevabını yazınız. Acaba verimizin kalitesi, ön işlem yapmadan çalışmaya uygun mudur? Neden bu kanıya vardınız?
Histogram bize standart bir dağılım örüntüsü verdi Kutu grafiklerinde ortanca değerler çok benzer Kümeleme analizi sonucunda, kontrol ve hastalıklı örnekler ayrı gruplandı
29.3 Ayrımsal gen anlatım analizi
Şimdi de bütün genler için ayrımsal gen anlatım analizi yapınız:
# once p degerlerini toplayacagimiz bir boş vektor oluşturalim
p.value <- rep(0, nrow(microarray))
for (i in 1:nrow(microarray)){
kontrol <- microarray[i, metadata$Durum=="normal"]
uygulama <- microarray[i, metadata$Durum=="glioblastoma"]
test <- t.test(x = kontrol, y = uygulama)
p.value[i] <- test$p.value
}Kaç tane gen anlamlı değişmiş? (0.001’e göre):
sum(p.value < 0.001, na.rm = TRUE)[1] 400Şimdi de anlatımları en farklı değişmiş bir tane geni ineceleyelim. Bu gen kaçıncı sırada?
which.min(p.value)[1] 305Bu genin kutu grafiğine bakalım mı?
boxplot(microarray[305,]~metadata$Durum)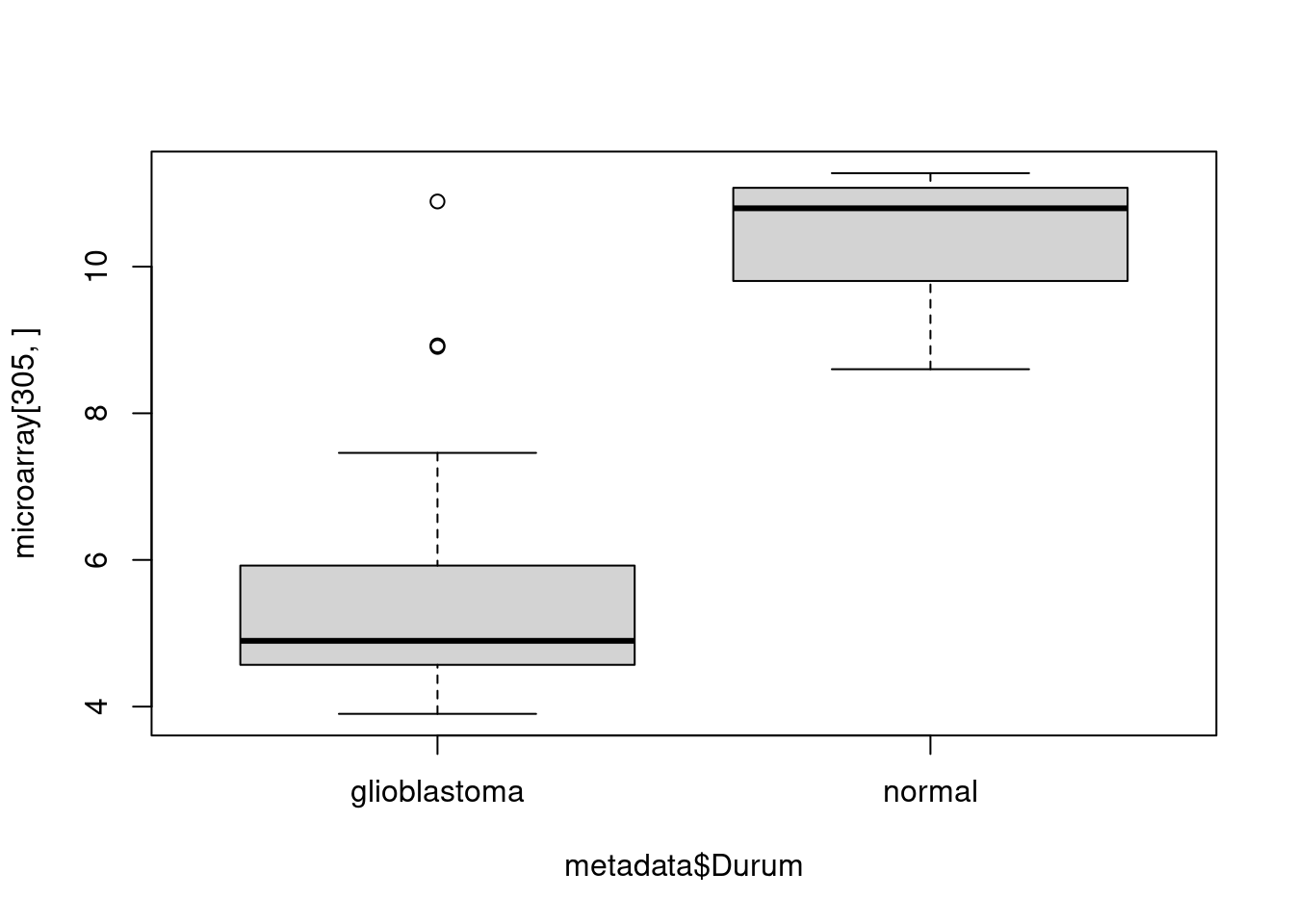
Bu genin anlatımı, glibolastoma koşulunda normal koşula göre nasıl değişmiş? Cevabı aşağıya yazınız:
Normal koşula göre azalmış